


 Sign Out
Sign Out

Pharmacology: Pharmacodynamics: Divalproex sodium is a stable coordination compound comprised of sodium valproate and valproic acid in a 1:1 molar relationship and formed during the partial neutralization of valproic acid with 0.5 equivalent of sodium hydroxide. Divalproex sodium is chemically designated as sodium hydrogen bis (2propylpentanoate). Divalproex sodium has a molecular weight of 310.41 and occurs as a white powder with a characteristic odor. Its empirical formula is C16H31NaO4.
Mechanism of action and Pharmacodynamic properties: Divalproex sodium dissociates to the valproate ion in the gastrointestinal tract. The mechanisms by which valproate exerts its therapeutic effects have not been established. It has been suggested that its activity in epilepsy is related to increased brain concentrations of gamma-aminobutyric acid (GABA).
Description of clinical studies: Epilepsy Complex Partial Seizures (CPS): The studies described in the following section were conducted using divalproex sodium tablets.
The efficacy of divalproex sodium in reducing the incidence of complex partial seizures (CPS) that occur in isolation or in association with other seizure types was established in two controlled trials.
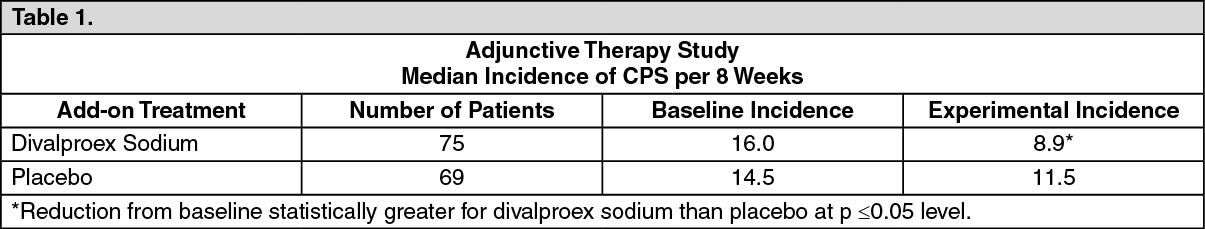
In one, multiclinic, placebo controlled study employing an add-on design (adjunctive therapy), 144 patients who continued to suffer eight or more CPS per eight weeks during an 8-week period of monotherapy with doses of either carbamazepine or phenytoin sufficient to assure plasma concentrations within the "therapeutic range," were randomized to receive, in addition to their original antiepilepsy drug (AED), either divalproex sodium or placebo. Randomized patients were to be followed for a total of 16 weeks. Table 1 presents the findings. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageFigure 1 presents the proportion of patients (X-axis) whose percentage reduction from baseline in complex partial seizure rates was at least as great as that indicated on the Y-axis in the adjunctive therapy study. A positive percent reduction indicates an improvement (i.e., a decrease in seizure frequency), while a negative percent reduction indicates worsening. Thus, in a display of this type, the curve for an effective treatment is shifted to the left of the curve for placebo. This figure shows that the proportion of patients achieving any particular level of improvement was consistently higher for divalproex sodium than for placebo. For example, 45% of patients treated with divalproex sodium had a ≥ 50% reduction in complex partial seizure rate compared to 23% of patients treated with placebo. (See Figure 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe second study assessed the capacity of divalproex sodium to reduce the incidence of CPS when administered as the sole AED. The study compared the incidence of CPS among patients randomized to either a high or low dose treatment arm. Patients qualified for entry into the randomized comparison phase of this study only if: 1) they continued to experience two or more CPS per four weeks during an 8 to 12 week long period of monotherapy with adequate doses of an AED (i.e., phenytoin, carbamazepine, phenobarbital, or primidone); and 2) they made a successful transition over a two week interval to divalproex sodium. Patients entering the randomized phase were then brought to their assigned target dose, gradually tapered off their concomitant AED and followed for an interval as long as 22 weeks. Less than 50% of the patients randomized, however, completed the study. In patients converted to divalproex sodium monotherapy, the mean total valproate concentrations during monotherapy were 71 and 123 mcg/mL in the low dose and high dose groups, respectively.
Table 2 presents the findings for all patients randomized who had at least one post-randomization assessment. (See Table 2.)
 Click on icon to see table/diagram/image
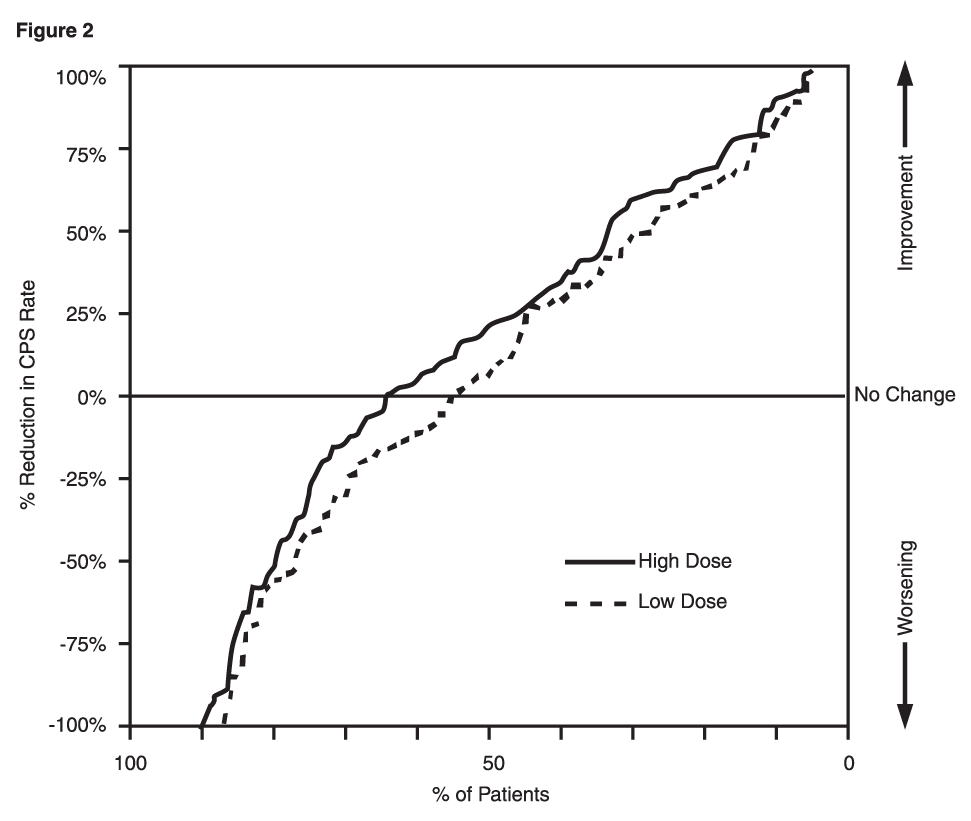
Click on icon to see table/diagram/imageFigure 2 presents the proportion of patients (X-axis) whose percentage reduction from baseline in complex partial seizure rates was at least as great as that indicated on the Y-axis in the monotherapy study. A positive percent reduction indicates an improvement (i.e., a decrease in seizure frequency), while a negative percent reduction indicates worsening. Thus, in a display of this type, the curve for a more effective treatment is shifted to the left of the curve for a less effective treatment. This figure shows that the proportion of patients achieving any particular level of reduction was consistently higher for high dose divalproex sodium than for low dose divalproex sodium. For example, when switching from carbamazepine, phenytoin, phenobarbital or primidone monotherapy to high dose divalproex sodium monotherapy, 63% of patients experienced no change or a reduction in complex partial seizure rates compared to 54% of patients receiving low dose divalproex sodium. (See Figure 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn a clinical trial of divalproex sodium as monotherapy in patients with epilepsy, 34/126 patients (27%) receiving approximately 50 mg/kg/day on average, had at least one value of platelets ≤ 75 x 109/L. Approximately half of these patients had treatment discontinued, with return of platelet counts to normal. In the remaining patients, platelet counts normalized with continued treatment. In this study, the probability of thrombocytopenia appeared to increase significantly at total valproate concentrations of ≥ 110 mcg/mL (females) or ≥ 135 mcg/mL (males).
In a double-blind, multicenter trial of valproate in elderly patients with dementia (mean age was 83 years old), doses were increased by 125 mg/day to a target dose of 20 mg/kg/day. A significantly higher proportion of valproate patients had somnolence compared to placebo, and although not statistically significant, there was a higher proportion of patients with dehydration. Discontinuations for somnolence were also significantly higher than with placebo. In some patients with somnolence (approximately one-half), there was associated reduced nutritional intake and weight loss. There was a trend for the patients who experienced these events to have a lower baseline albumin concentration, lower valproate clearance, and a higher BUN.
Mania: The effectiveness of divalproex sodium extended-release tablets for the treatment of acute mania is based in part on studies establishing the effectiveness of divalproex sodium delayed release tablets for this indication. Divalproex sodium extended-release tablets' effectiveness was confirmed in one randomized, double-blind, placebo-controlled, parallel group, 3-week, multicenter study. The study was designed to evaluate the safety and efficacy of divalproex sodium extended-release tablets in the treatment of bipolar I disorder, manic or mixed type, in adults. Adult male and female patients who had a current DSM-IV TR primary diagnosis of bipolar I disorder, manic or mixed type, and who were hospitalized for acute mania, were enrolled into this study. Divalproex sodium extended-release tablets were initiated at a dose of 25 mg/kg/day given once daily, increased by 500 mg/day on Day 3, then adjusted to achieve plasma valproate concentrations in the range of 85-125 mcg/mL.
Mean daily divalproex sodium extended-release tablets doses for observed cases were 2,362 mg (range: 500-4,000), 2,874 mg (range: 1,500-4,500), 2,993 mg (range: 1,500-4,500), 3,181 mg (range: 1,500-5,000), and 3,353 mg (range: 1,500-5,500) at Days 1, 5, 10, 15 and 21, respectively. Mean valproate concentrations were 96.5 mcg/mL, 102.1 mcg/mL, 98.5 mcg/mL, 89.5 mcg/mL at Days 5, 10, 15 and 21, respectively. Patients were assessed on the Mania Rating Scale (MRS; score ranges from 0-52).
Divalproex sodium extended-release tablets were significantly more effective than placebo in reduction of the MRS total score.
Migraine: The results of a multicenter, randomized, double-blind, placebo-controlled, parallel-group clinical trial demonstrated the effectiveness of divalproex sodium extended-release in the prophylactic treatment of migraine headache. This trial recruited patients with a history of migraine headaches with or without aura occurring on average twice or more a month for the preceding three months. Patients with cluster or chronic daily headaches were excluded. Women of childbearing potential were allowed in the trial if they were deemed to be practicing an effective method of contraception.
Patients who experienced ≥ two migraine headaches in the four-week baseline period were randomized in a 1:1 ratio to divalproex sodium extended-release or placebo and treated for twelve weeks. Patients initiated treatment on 500 mg once daily for one week, and were then increased to 1,000 mg once daily with an option to permanently decrease the dose back to 500 mg once daily during the second week of treatment if intolerance occurred. Ninety-eight of 114 divalproex sodium extended-release-treated patients (86%) and 100 of 110 placebo-treated patients (91%) treated at least two weeks maintained the 1,000 mg once daily dose for the duration of their treatment periods.
Treatment outcome was assessed on the basis of reduction in four-week migraine headache rate in the treatment period compared to the baseline period.
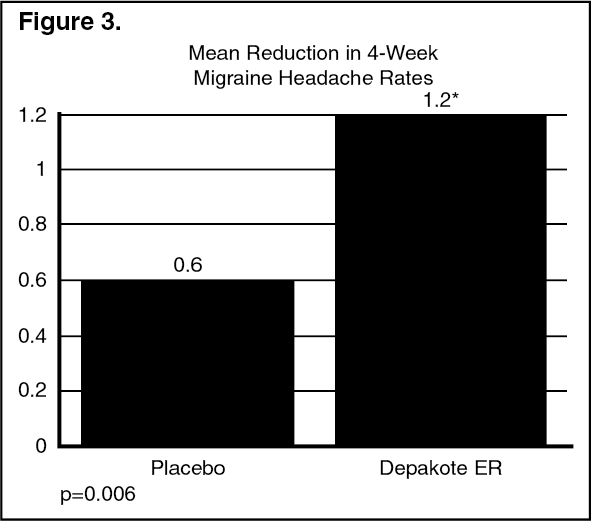
Patients (50 male, 187 female) ranging in age from 16 to 69 were treated with divalproex sodium extended-release (n=122) or placebo (n=115). Four patients were below the age of 18 and three were above the age of 65. Two hundred and two patients (101 in each treatment group) completed the treatment period. The mean reduction in four-week migraine headache rate was 1.2 from a baseline mean of Precautions in the divalproex sodium extended-release group, versus 0.6 from a baseline mean of Dosage & Administration in the placebo group. The treatment difference was statistically significant (see Figure 3).
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePharmacokinetics: Absorption/Bioavailability: The absolute bioavailability of divalproex sodium extended-release (ER) tablets, administered as a single dose after a meal, was approximately 90% relative to intravenous infusion.
When given in equal total daily doses, the bioavailability of divalproex sodium ER is less than that of divalproex sodium (divalproex sodium enteric-coated tablets). In five multiple-dose studies in healthy subjects (n=82) and in subjects with epilepsy (n=86), when administered under fasting and nonfasting conditions, divalproex sodium ER given once daily produced an average bioavailability of 89% relative to an equal total daily dose of divalproex sodium given b.i.d., t.i.d., or q.i.d. The median time to maximum plasma valproate concentrations (Cmax) after divalproex sodium ER administration ranged from 4 to 17 hours. After multiple once daily dosing of divalproex sodium ER, the peak-to-trough fluctuation in plasma valproate concentrations was 10-20% lower than that of regular divalproex sodium given b.i.d., t.i.d., or q.i.d.
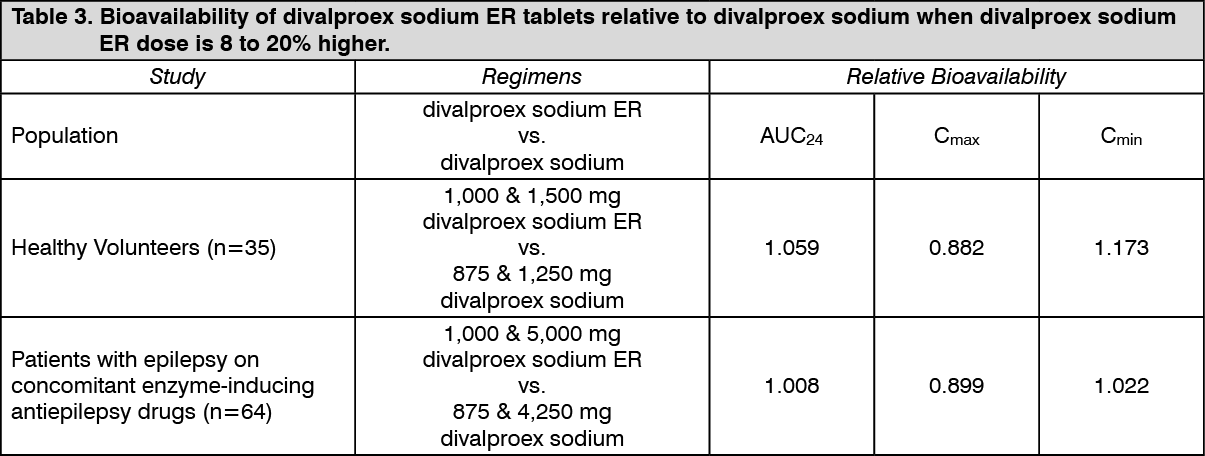
Conversion from divalproex sodium to divalproex sodium ER: When divalproex sodium ER is given in doses 8 to 20% higher than the total daily dose of divalproex sodium, the two formulations are bioequivalent. In two randomized, crossover studies, multiple daily doses of divalproex sodium were compared to 8 to 20% higher once-daily doses of divalproex sodium ER. In these two studies, divalproex sodium ER and divalproex sodium regimens were equivalent with respect to area under the curve (AUC; a measure of the extent of bioavailability). Additionally, valproate Cmax was lower, and Cmin was either higher or not different, for divalproex sodium ER relative to divalproex sodium regimens (see following Table 3). (See Table 3.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageConcomitant antiepilepsy drugs (topiramate, phenobarbital, carbamazepine, phenytoin, and lamotrigine were evaluated) that induce the cytochrome P450 isozyme system did not significantly alter valproate bioavailability when converting between divalproex sodium and divalproex sodium ER.
Distribution: Protein Binding: The plasma protein binding of valproate is concentration dependent and the free fraction increases from approximately 10% at 40 mcg/mL to 18.5% at 130 mcg/mL. Protein binding of valproate is reduced in the elderly, in patients with chronic hepatic diseases, in patients with renal impairment, and in the presence of other drugs (e.g., aspirin). Conversely, valproate may displace certain protein-bound drugs (e.g., phenytoin, carbamazepine, warfarin, and tolbutamide) (see Interactions for more detailed information on the pharmacokinetic interactions of valproate with other drugs).
CNS Distribution: Valproate concentrations in cerebrospinal fluid (CSF) approximate unbound concentrations in plasma (about 10% of total concentration).
Placental transfer (see Use in Pregnancy & Lactation): Valproate crosses the placental barrier in animal species and in humans: In animal species, valproate crosses the placenta, to a similar extent as in humans.
In humans, several publications assessed the concentration of valproate in the umbilical cord of neonates at delivery. Valproate serum concentration in the umbilical cord, representing that in the fetuses, was similar to or slightly higher than that in the mothers.
Metabolism: Valproate is metabolized almost entirely by the liver. In adult patients on monotherapy, 30 to 50% of an administered dose appears in urine as a glucuronide conjugate. Mitochondrial β-oxidation is the other major metabolic pathway, typically accounting for over 40% of the dose. Usually, less than 15 to 20% of the dose is eliminated by other oxidative mechanisms. Less than 3% of an administered dose is excreted unchanged in urine.
The relationship between dose and total valproate concentration is nonlinear; concentration does not increase proportionally with the dose, but rather, increases to a lesser extent due to saturable plasma protein binding. The kinetics of unbound drug are linear.
Excretion: Mean plasma clearance and volume of distribution for total valproate are 0.56 L/hr/1.73 m2 and 11 L/1.73 m2, respectively. Mean plasma clearance and volume of distribution for free valproate are 4.6 L/hr/1.73 m2 and 92 L/1.73 m2. Mean terminal half-life for valproate monotherapy ranged from 9 to 16 hours following oral dosing regimens of 250 to 1,000 mg.
The estimates cited apply primarily to patients who are not taking drugs that affect hepatic metabolizing enzyme systems. For example, patients taking enzyme-inducing antiepileptic drugs (carbamazepine, phenytoin, and phenobarbital) will clear valproate more rapidly. Because of these changes in valproate clearance, monitoring of antiepileptic concentrations should be intensified whenever concomitant antiepileptics are introduced or withdrawn.
Special Populations: Neonates: In neonates and infants up to 2 months of age, valproate clearance is decreased when compared to adults. This is a result of reduced clearance (perhaps due to delay in development of glucuronosyltransferase and other enzyme systems involved in valproate elimination) as well as increased volume of distribution (in part due to decreased plasma protein binding). For example, in one study, the half-life in children under 10 days ranged from 10 to 67 hours compared to a range of 7 to 13 hours in children greater than two months.
Safety and effectiveness of divalproex sodium extended-release in the prophylaxis of migraine in pediatric patients have not been established. Therefore, the information as follows is applicable to the pediatric population when used for epilepsy indication only.
Geriatric: The capacity of elderly patients (age range: 68 to 89 years) to eliminate valproate has been shown to be reduced compared to younger adults (age range: 22 to 26). Intrinsic clearance is reduced by 39%; the free fraction of valproate is increased by 44%. Accordingly, the initial dosage should be reduced in the elderly (see Dosage & Administration).
Pediatric: Pediatric patients (i.e., between 3 months and 10 years) have 50% higher clearances expressed on weight (i.e., mL/min/kg) than do adults.
Above the age of 10 years, children and adolescents have valproate clearances similar to those reported in adults.
Based on published literature, in pediatric patients below the age of 10 years, the systemic clearance of valproate varies with age.
In children aged 2-10 years, valproate clearance is 50% higher than in adults.
The valproate pharmacokinetic profile following administration of divalproex sodium ER was characterized in a multiple-dose, non-fasting, open-label, multi-center study in children and adolescents. Divalproex sodium ER once-daily doses ranged from 250 to 1,750 mg. Once-daily administration of divalproex sodium ER in pediatric patients (10-17 years) produced plasma valproic acid concentration-time profiles similar to those that have been observed in adults.
Safety and effectiveness of divalproex sodium extended-release in the prophylaxis of migraine in pediatric patients have not been established. Therefore, the information as follows is applicable to the pediatric population when used for epilepsy indication only.
Gender: There are no differences in the body surface area adjusted unbound clearance between males and females (4.8±0.17 and 4.7±0.07 L/hr per 1.73 m2, respectively).
Ethnicity: The effects of ethnicity on the kinetics of valproate have not been studied.
Renal impairment: A slight reduction (27%) in the clearance of unbound valproate has been reported in patients with renal failure (creatinine clearance < 10 mL/minute); however, hemodialysis typically reduces valproate concentrations by about 20%. Protein binding in these patients is substantially reduced; thus, monitoring total concentrations may be misleading. For further guidance refer to Dosage & Administration.
Hepatic impairment: See Contraindications and Hepatotoxicity/Hepatic dysfunction under Precautions.
Liver disease impairs the capacity to eliminate valproate. In one study, the clearance of free valproate was decreased by 50% in seven patients with cirrhosis and by 16% in four patients with acute hepatitis, compared to six healthy subjects. In that study, the half-life of valproate was increased from 12 to 18 hours. Liver disease is also associated with decreased albumin concentrations and larger unbound fractions (2 to 2.6 fold increase) of valproate. Accordingly, monitoring of total concentrations may be misleading since free concentrations may be substantially elevated in patients with hepatic disease whereas total concentrations may appear to be normal.
Plasma Levels and Clinical Effect: The relationship between plasma concentration and clinical response is not well documented. One contributing factor is the nonlinear, concentration dependent protein binding of valproate that affects the clearance of the drug. Thus, monitoring of total serum valproate cannot provide a reliable index of the bioactive valproate species.
For example, because the plasma protein binding of valproate is concentration dependent, the free fraction increases from approximately 10% at 40 mcg/mL to 18.5% at 130 mcg/mL. Higher than expected free fractions occur in the elderly, in hyperlipidemic patients, and in patients with hepatic and renal diseases.
Mania: In a placebo-controlled clinical trial of acute mania, patients were dosed to clinical response with trough plasma concentrations between 85 and 125 mcg/mL.
Epilepsy: The therapeutic range in epilepsy is commonly considered to be 50 to 100 mcg/mL of total valproate, although some patients may be controlled with lower or higher plasma concentrations.
Equivalent doses of valproate sodium and divalproex sodium yield equivalent plasma levels of the valproate ion.
Toxicology: Preclinical safety data: Carcinogenesis, Mutagenesis, Reproductive and Developmental Toxicity and Impairment of Fertility: Carcinogenesis: The 2-year carcinogenicity studies were conducted in mice and rats given oral valproate doses of approximately 80 and 160 mg/kg/day (which are the maximum tolerated doses in these species but less than the maximum recommended human dose based on body surface area). Subcutaneous fibrosarcomas were observed in male rats and hepatocellular carcinomas and bronchiolo-alveolar adenomas were observed in male mice at incidences slightly higher than concurrent study controls but comparable to historical control data.
Mutagenesis: Valproate was not mutagenic in an in vitro bacterial assay (Ames test), did not produce dominant lethal effects in mice, and did not increase chromosome aberration frequency in an in vivo cytogenetic study in rats. Valproate was not mutagenic in bacteria (Ames test) or mouse lymphoma L5178Y cells at thymidine kinase locus (mouse lymphoma assay) and did not induce DNA repair activity in primary culture of rat hepatocytes. It did not induce either chromosome aberrations in rat bone marrow or dominant lethal effects in mice after oral administration. In literature, after intraperitoneal exposure to valproate, increased incidences of DNA and chromosome damage (DNA strand-breaks, chromosomal aberrations or micronuclei) have been reported in rodents. However, the relevance of the results obtained with the intraperitoneal route of administration is unknown. Statistically significant higher incidences of sister-chromatid exchange (SCE) have been observed in patients exposed to valproate as compared to healthy subjects not exposed to valproate. However, these data may have been impacted by confounding factors. Two published studies examining SCE frequency in epileptic patients treated with valproate versus untreated epileptic patients, provided contradictory results.
The biological significance of an increase in SCE frequency is not known.
Reproductive and Developmental Toxicity: Teratogenic effects (malformations of multiple organ systems) have been demonstrated in mice, rats, and rabbits.
In published literature, behavioral abnormalities have been reported in first generation offspring of mice and rats after in utero exposure to clinically relevant doses/exposures of valproate.
In mice, behavioral changes have also been observed in the 2nd and 3rd generations, albeit less pronounced in the 3rd generation, following an acute in utero exposure of the first generation. The relevance of these findings for humans is unknown.
Impairment of fertility: In sub-chronic/chronic toxicity studies, testicular degeneration/atrophy or spermatogenesis abnormalities and a decrease in testes weight were reported in adult rats and dogs after oral administration starting at doses of 1250 mg/kg/day and 150 mg/kg/day, respectively.
In a fertility study in rats, valproate at doses up to 350 mg/kg/day did not alter male reproductive performance.
In juvenile rats, a decrease in testes weight was only observed at doses exceeding the maximum tolerated dose (from 240 mg/kg/day by intraperitoneal or intravenous route) and with no associated histopathological changes. No effects on the male reproductive organs were noted at tolerated doses (up to 90 mg/kg/day). Relevance of the testicular findings to pediatric population is unknown.
However, male infertility has been identified as an undesirable effect in humans (see Use in Pregnancy & Lactation and Adverse Reactions).